

In the Medical Device industry software components, whether standalone or as part of a physical device, must follow the same rules as any other component, i.e. ISO 14971 “Medical devices – Application of risk management to medical devices”.
BUT

.. but there are significant deviations required by IEC 62304 and the GDA guidance document “Content of Premarket Submissions for Device Software Functions”
In particular:
It is often difficult to adequately estimate the probability of software failures that could contribute to a hazardous situation. Applying unrealistically low probability estimates to software failures could result in unrealistic risk evaluation and subsequently lead to inappropriate risk control measures. As a result, in some instances it may be prudent to focus on the identification of potential software functionality and failures that could result in hazardous situations instead of estimating probability. In such cases, considering a worse case probability is appropriate, the probability for the software failure occurring should be set to 1.
This is not a small demand. It compromises the typical approach to risk management, where controls and mitigations are used to reduce the probability of the hazardous situation occurring. No matter what you do, P=100% before and after mitigation. Even if you use a split probability P1 and P2, P = P1 x P2 = P2, and P2 is not usually modified, as it ties in with clinical effects.
In your risk model, the risk level will remain in the same range:

How is it possible then to set the acceptability of each individual risk item? How can a company determine if it has done enough?
There are two general approaches that have been accepted by the FDA and Notified Bodies, and they are described below.
Line-by-Line Justification
With this approach, the Product Development team must explain why the selected controls are sufficient for each risk and reduce the risk As-Low-As-Possible. This is a time-consuming task, where users are tempted to copy and paste the same justification over and over, defying the purpose of this assessment.
It is a safe approach though, as the Severity of the risk is the only parameter used to make the acceptability assessment.
The Third Parameter
The Development Team can also define an additional parameter, let’s call it a “mitigation level”, and assign a value based on the effectiveness of the controls the team thinks that they have.
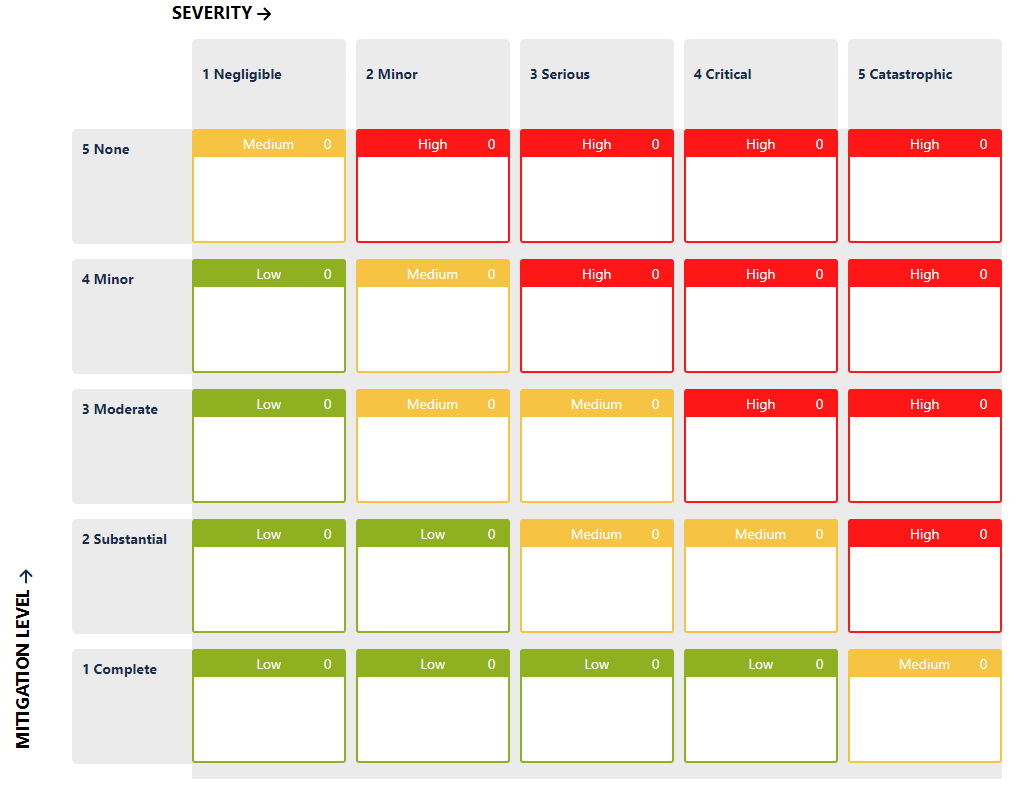
It may look like a Probability, it may work the same as Probability, but it’s not Probability.
Please note that these levels, their names and descriptions are arbitrary.
… and more
If deemed necessary, the team can also define the mitigation level as a function of other factors, for example the type of mitigation:
| Type of Mitigation | Mitigation Level |
|---|---|
| None | None |
| Software, in the same component as the one being assessed | Minor |
| Information (e.g. training, manual) | Minor |
| Software, in a different software system as the one being assessed (e.g. a watchdog) | Moderate |
| Protection (e.g. alarms) | Moderate |
| Design | High |
Please note that these levels, their names and descriptions are arbitrary.
Conclusion
Despite losing a valuable parameter such as Probability, there are several solutions that a development team can adopt to assess the risk level and acceptability for software components or products.
Just do not use the word “Probability”.
For more information about Medical Device Risk Management app in Atlassian Jira, check out the most advanced Risk Management app in Jira – you can try it out for free for 30 days. Alternatively, schedule a call with the SoftComply team!
Follow us on LinkedIn for more on Risk Management:


