

Intro
FDA aims to provide a regulatory framework that supports innovation in medical device software development. As such, they understand the need to rapidly change and update software without affecting the safety and effectiveness of the device. In such cases, a new submission shouldn’t always required. But when?
Overview
In August 2024 the FDA release a new Draft Guidance “Predetermined Change Control Plans for Medical Devices” (PCCP), shortly followed in December 2024 by “Marketing Submission Recommendations for a Predetermined Change Control Plan for Artificial Intelligence-Enabled Device Software Functions”.
They stem from the same principles of certain sections of the existing guidance documents: “Deciding When to Submit a 510(k) for a Change to an Existing Device” and “Replacement Reagent and Instrument Family Policy for In Vitro Diagnostic Devices”. But while the existing documents describe when a new submission is required, leaving the manufacturer to decide whether the changes meet the criteria (often quite a grey area), the new guidance documents propose the use of the PCCP process, where the manufacturer agrees upfront with the FDA which changes do not require a new submission.
In addition, the PCCP must also include the description of what the manufacturer will do when these changes are implemented. The following diagram, taken from the first guidance, explains visually and in simple terms what the process looks like:
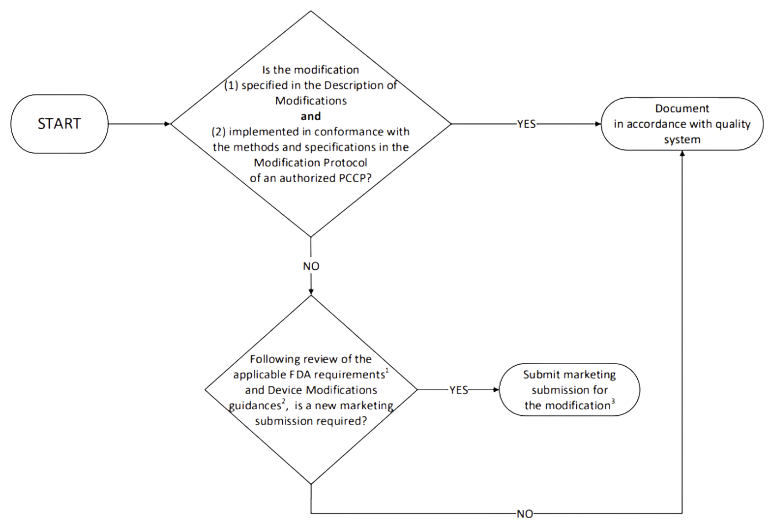
Content of the PCCP
1. Description of each modification with sufficient details to prevent subjective interpretation and confusion. Modifications must be presented at a level of detail that permits understanding of the specific modifications that will be made to the device. NOTE: the FDA guidance documents provide example and directions on what type of changes, in general, may be part of a PCCP and which ones should not.
2. Description of how each modification is implemented, focusing on the change management process for the development, verification, validation, documentation requirements, (re)training and more.
The FDA expects to see well defined V&V process, protocols, acceptance criteria, including Performance Evaluation Methods and Update Procedures.
3. Traceability between the Description of Modifications section and the Modification Protocol section, where every proposed modification is covered in both Performance Evaluation Methods and Update Procedures sections.
4. Impact Assessment covering “the benefits and risks of implementing a PCCP for a device, as well as the mitigations of those risks”. In practice it is a pre-modification risk management activity, including resulting benefit/risk analysis for each modification and all modifications cumulatively. This means that the modifications must be sufficiently detailed to be comprehensively analysed for failures, effects and controls (these controls must be applied as part of the modification).
PCCP for AI-enabled devices
A PCCP for a device including AI-enabled functions follows the same structure as the one previously described, but requires additional AI-specific details:
1. Description of each modification:
- State if the planned modifications are proposed to be implemented automatically, manually, or a combination of both. It may be helpful for manufacturers to clearly establish boundaries or guardrails that define the range of automatic updates.
- Clearly specify if the proposed modifications will be implemented in a uniform manner across all devices on the market, and/or implemented differently on different devices.
- The expected frequency of updates.
- Labeling sections that are anticipated to be impacted for each local adaptation.
2. How each modification is implemented:
- data management practices;
- re-training practices;
- performance evaluation protocols, and
- update procedure.
3. Traceability. Each modification must be traceable to the relevant portion of each of the 4 items above.
4. Impact Assessment. Include how the modification(s) impact other device software functions and/or device hardware (if applicable).
Changes to an authorized PCCP
As always, not all changes can be foreseen by the manufacturer at the launch of their product.
The FDA accepts that an authorized PCCP may need to be updated several times during the life of a device, but “FDA believes that modifications to an authorized PCCP will generally constitute changes to the device that would otherwise require a new marketing submission”. The guidance documents list what types of PMA supplement / 510(k) submission may be appropriate in these cases.
It is still a submission, but it focuses on a single document rather than a full DHF.
Note that this also applies to new PCCPs submitted for existing authorized devices.
Conclusion
In the world of regulatory compliance, a well-crafted Predetermined Change Control Plan (PCCP) isn’t just a nice-to-have — it’s a necessity. By proactively managing changes, manufacturers can stay ahead of the curve, ensuring their AI-enabled devices evolve in line with FDA expectations and maintain the trust of both regulators and patients.
How can SoftComply help?
SoftComply’s solutions can play a crucial role in helping companies implement and manage PCCPs effectively. The SoftComply Document Manager is essential for maintaining PCCP documentation in compliance with FDA requirements, while the SoftComply Risk Manager Plus is an ideal solution for managing the risk assessment aspects of PCCPs.
SoftComply apps are built on Atlassian Jira and Confluence providing a seamless environment for managing all aspects of PCCPs, from risk assessment to documentation, making it an invaluable asset for companies navigating the complex landscape of AI-enabled medical device regulation.
![]() In our recent webinar we talked about FDA guidance on AI-enabled medical devices and PCCP – have a listen to the discussion:
In our recent webinar we talked about FDA guidance on AI-enabled medical devices and PCCP – have a listen to the discussion:



